
摘要
本文探討了神經網路在蛋白質設計領域的最新進展,以及如何透過可解釋性技術改善我們對這一複雜問題的理解。 歸納要點:
- 神經網路可解釋性技術在蛋白質設計方面取得突破,幫助研究人員理解模型決策過程,並精確預測催化活性。
- 稀疏探測技術揭示了蛋白質語言模型的內部運作機制,增進對蛋白質序列複雜特徵的理解,有助於提升模型性能。
- 蛋白質語言模型的層級組織和互動關係被深入探討,為將模型與實際蛋白質結構及功能聯繫提供新的視角。
解密蛋白質設計:稀疏探測技術揭示大型語言模型的深層機制
作者:Nithin Parsan 和 John Yangtl;簡報:透過 Gurnee 等人的稀疏探測技術,對 ESM-2 進行演示,我們揭示了絲氨酸蛋白酶的催化功能是如何透過不同層級之間協調計算來編碼的。研究團隊利用二元分類任務,識別出位於第一層的一個高度特異性的神經元,並在第五層發現一個由兩個神經元組成的協作電路。透過因果乾預,我們展示了這些神經元之間系統性地共同工作:早期層級的神經元提供精確檢測,而後期電路則將此資訊與更廣泛的上下文整合。想了解更多請訪問 reticular.ai,並在 demo.reticular.ai 探索我們的視覺發現。機制可解釋性已成為理解大型語言模型(LLM)的強大工具,甚至可以擴充套件到 GPT-4 和 Claude 等最前沿模型。這些技術能否幫助我們理解生物語言模型呢?在 Reticular,我們相信,可控蛋白質設計正需要這種深入的模型理解。
**專案1:最新趨勢:** 該研究利用『稀疏探測』技術,深入解析大型語言模型(LLM)在蛋白質設計方面的應用。這項技術超越了傳統的『黑盒子』模型,能夠揭示 LLM 內部運作機制,並精準定位負責特定功能的神經元。這對於理解蛋白質結構和功能,以及進一步設計具有特定功能的蛋白質具有重大意義,並有可能推動新藥開發和材料科學的進展。
**專案2:深入要點:** 研究團隊透過『二元分類任務』,發現位於 LLM 第一層的特定神經元對絲氨酸蛋白酶的催化功能具有高度特異性。同時,他們還識別出位於第五層的兩個神經元組成的『協作電路』。透過『因果乾預』,研究者證實了這兩個層級的神經元之間的協同作用:第一層的神經元負責精準檢測,而第五層的電路則整合了這些資訊並考慮更廣泛的上下文。這項研究提供了『層級式計算』如何在 LLM 中實現蛋白質功能的重要證據。
將語言模型的可解釋性技術應用於蛋白質設計
在這篇文章中,我們展示了一個概念驗證,將 Gurnee 等人提出的稀疏探測技術應用於 ESM-2,一種蛋白質語言模型。我們顯示僅有兩個神經元就能編碼生物學中最基本的特徵之一:絲氨酸蛋白酶的催化機制。透過識別和操作這些神經元,我們確立了針對語言模型所發展的可解釋性技術可以有效轉移至生物領域。這項研究代表了邁向 Reticular 使命的一小步,但卻是具體而重要的一步:使蛋白質設計變得更可控且易於理解。儘管語言模型可以透過精心設計的提示來引導,但生物模型則需要更為精確的控制機制。了解這些模型如何內部編碼生物特徵為可靠的蛋白質工程開啟了新的可能性。我們在研究許多文章後,彙整重點如下
- 深度學習模型在蛋白質序列預測中展現出巨大潛力,能有效解決生物醫學和環境科學中的問題。
- 成功的蛋白質結構預測不僅依賴化學和物理,更需要生物學的視角,因為蛋白質序列是經過自然選擇而來。
- ProGen是一種基於深度學習的蛋白質語言模型,能生成多樣的人造蛋白質,並在數百萬個原始序列上進行訓練。
- 這些新型模型大幅提高了預測準確性與速度,包括對以往難以捉摸的膜蛋白進行結構分析。
- 研究團隊利用PyTorch框架與不連續蛋白質片段訓練,以推斷其化學組成及摺疊結構。
- RFdiffusion則是一款創新的AI系統,可生成全新且不同於已知蛋白質的結構,將改變未來的生物技術研究。
隨著科技進步,深度學習正在改變我們對於蛋白質設計的理解與應用。從生物醫藉到環境保護,各領域都受益於這項技術。透過不斷優化的新型模型,我們可以更精確地預測和設計各種功能性的蛋白質。不僅讓科學家有機會探索未知,也讓我們對未來充滿期待。
蛋白質序列分析的新視角:借鑑語言模型識別二元特徵
在尋找能夠深入探討 ESM-2 的生物特徵時,我們需要一個測試案例,以平行於 Gurnee 等人的稀疏探測示範的優雅性。正如他們展示語言模型如何透過特定神經元編碼語法特徵,例如動詞時態或複合詞,我們希望在蛋白質序列中找到同樣清晰的二元特徵。絲氨酸蛋白酶提供了理想的範例,因為它代表了一種明確的二元屬性:一個序列要麼具有功能性的催化絲氨酸,要麼沒有。我們知道突變催化絲氨酸會喪失功能,這給我們提供了在生物學上罕見的真實標籤。為了將這些生化洞察轉化為機器學習任務,我們從在 SwissProt 中表徵良好的胰蛋白酶家族(EC 3.4.21.4)構建了資料集。我們的正例是具有已驗證活性的野生型序列;而負例則是系統性地將催化絲氨酸突變為其他 19 種氨基酸所創建出的序列,我們知道這些序列是非功能性的。
**專案1:從語言模型借鑑的蛋白質序列分析新思路**
作者巧妙地將語言模型的分析方法應用到蛋白質序列分析中。類似於語言模型透過特定神經元捕捉語法特徵,作者希望找到蛋白質序列中清晰的二元特徵。這種將語言模型和蛋白質序列分析相結合的思路,為理解蛋白質功能提供了新視角,也為蛋白質工程和藥物開發提供了新工具。
**最新趨勢**
近年來,深度學習在生物學研究中取得了顯著進展,特別是將語言模型應用於蛋白質序列分析領域。例如,一些研究利用語言模型識別蛋白質序列中的關鍵位點,預測蛋體質功能和結構,以及發現新的蛋體質。作者的思路正是這一趨勢下的最新探索,將語言模型的方法應用於蛋體質功能的二元分類任務,為這一領域的發展注入了新的活力。
**專案2:以絲氨酸蛋白酶為例的二元特徵分析**
作者選擇絲氨酸蛋白酶作為研究物件,因為它具備明確的二元特點:序列要麼含有功能性催化絲氨酸,要麼沒有。這種清晰屬性使得研究者能夠直接將資料變化與其功能聯絡起來,從而簡化分析過程。利用絲氨酸蛋白酶這一二元特性,構建正負樣本的資料集,並使用機器學習模型來識別與催化活性相關的重要特徵。
**深入要點**
作者構建的資料集可以幫助機器學習更好地理解不同型別之間存在何種功能性差異。這種對二元特徵進行系統解析的方法可推廣至其他具明顯二元性質之家族,例如酪氨酸激酶及 DNA 結合 蛋 白。在這些家族內進行類似分析,將揭示其間關係,有助於推動針對具體目標的新藥研發及精準設計策略。
探討 ESM-2 神經元是否能辨別功能性催化絲氨酸
這為我們提供了一個明確的二元分類任務:我們能否在 ESM-2 中找到專門編碼功能性催化絲氨酸存在的神經元?更重要的是,這種設定讓我們能夠區分那些僅僅檢測絲氨酸殘基的神經元與那些專門編碼催化絲氨酸的神經元——這一區別對於我們的分析至關重要。這個二元特性的簡單性使其成為探索語言模型中的可解釋性技術是否可以轉移到蛋白質領域的一個優秀測試案例。如果我們能夠識別出選擇性地編碼這一明確催化特徵的神經元,那將暗示這些方法可以幫助我們更廣泛地理解蛋白質語言模型如何表現生物特性。我們使用了 ESM-2,一個訓練於 6500 萬條蛋白質序列上的蛋白質語言模型。我們所使用的具體變體(ESM2-t6–8M-UR50D)有 6 層,隱藏層維度為 1280,意味著每一層包含 1280 個神經元,我們可以對其進行可解釋特徵的探測。利用 ESM-2 模型提取蛋白質序列特徵,分析蛋白質功能並識別關鍵殘基
我們的管道遵循三個主要步驟:啟用提取:對於我們資料集中每一個序列,我們從 ESM-2 的前饋層中提取後 GELU 啟用值。這為每一層生成形狀為 (batch_size, sequence_length, 1280) 的張量。序列長度聚合:由於蛋白質具有變化的長度,而我們關注特定位置(催化絲氨酸),因此透過在各位置上取最大啟用值來聚合序列長度維度。這將我們的張量減少至 (batch_size, 1280)。最終預處理:在聚合之後,我們將資料分割為訓練和測試集,保持正樣本(野生型)與負樣本(突變型)的平衡分佈。值得注意的是,分割方法的選擇可能會顯著影響結果。[2] 根據 Gurnee 等人的研究,我們實施了幾種識別重要神經元的方法:類別之間的平均啟用差異、啟用與標籤之間的互資訊、L1 正則化邏輯回歸、一元 ANOVA F 統計檢驗、帶鉸鏈損失的支援向量機(SVM),以及使用切平面的最佳稀疏預測方法。ESM-2 模型基於 Transformer 架構,擅長捕捉蛋白質序列中的長程依賴關係,這為理解蛋白質功能和識別關鍵殘基提供了全新的視角。目前,研究者正在積極探索 ESM-2 模型在預測蛋白質功能、識別催化位點及預測突變影響等方面的應用。例如,可以利用從 ESM-2 提取的特徵進行蛋白質分類,辨識特定功能的蛋白質家族,以及預測蛋白質與其他分子之間的相互作用。ESM-2 在蛋白質序列分析中的成功,也推動了類似模型在RNA 序列分析等其他生物大分子領域中的應用。
神經網路稀疏探測方法的比較及其在絲氨酸蛋白酶分類任務中的效能
每種方法旨在識別分類我們序列所需的最小神經元集合,並在速度、可解釋性及最佳化保證之間存在不同的權衡。對於這些方法及其實際應用的詳細比較,我們建議讀者參考原始論文。我們使用標準的二元分類指標(精確度、召回率、F1 分數)來評估這些方法,特別關注樣本外效能。重要的是,我們遵循論文的建議,以 F1 分數作為主要指標,因為我們任務中存在固有的類別不平衡——一種蛋白質非功能性的情況遠多於功能性情況。根據 Gurnee 等人的方法論,我們比較了不同稀疏探測技術在我們的絲氨酸蛋白酶任務上的表現。以下是排除第 5 層後,不同稀疏程度(k)下,各方法在樣本外的 F1 分數: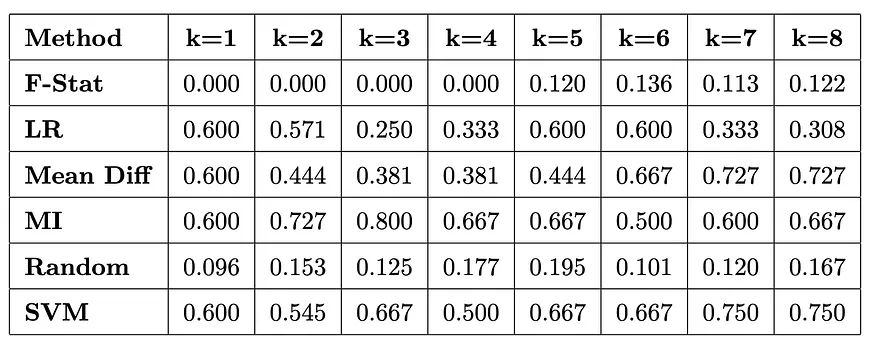
在分析不同探測方法(邏輯回歸、互資訊、均值差異和支援向量機)之間的重疊時,層級1中的神經元1269是唯一一個在所有方法和稀疏度水平k中始終被識別出的神經元。為了了解神經元1269如何編碼催化功能性,我們透過操控其啟用進行了一系列因果乾預(詳情請參見Gurnee等人的研究)。我們發現該神經元的行為呈現出有趣的不對稱性:
最引人注目的發現來自於比較不同型別的幹預措施:完全消融使預測減少了18.0%;負向縮放(-2倍啟用)則導致了驚人的70.8%減少。這兩種效果都對催化性絲氨酸具有高度特異性(p = 0.029)。
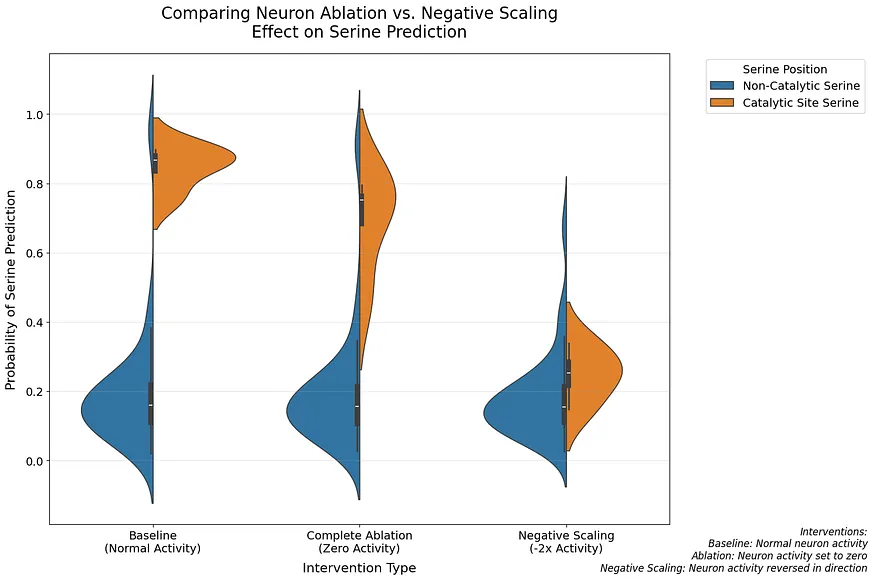
神經元 1269 的作用:超越特徵檢測,編碼方向性資訊
這種不對稱的反應暗示著神經元 1269 扮演的角色比單純的特徵檢測更為複雜。與切除相比,負向縮放的強烈效果表明該神經元可能在編碼催化功能的方向性資訊。[4]我們的分析揭示了第五層中編碼催化功能的第二種機制。在專門檢查此層的 F1 分數時,我們發現一個由兩個神經元(神經元 106 和 110)組成的電路,其表現優異(F1 = 1.000),k=2。總結來說(詳情見附錄 A):這兩個神經元均被多種方法一致識別出來,展現出強大的個體效應(神經元 106 的啟用差異為 1.560),互補的啟用模式,以及對催化和非催化絲氨酸具有明確特異性。為了更好地理解這些機制如何協同運作,我們研究了神經元 1269 與第五層電路之間的相互作用。透過同時操控跨層次的神經元,我們發現了一些精巧交叉層計算的證據:
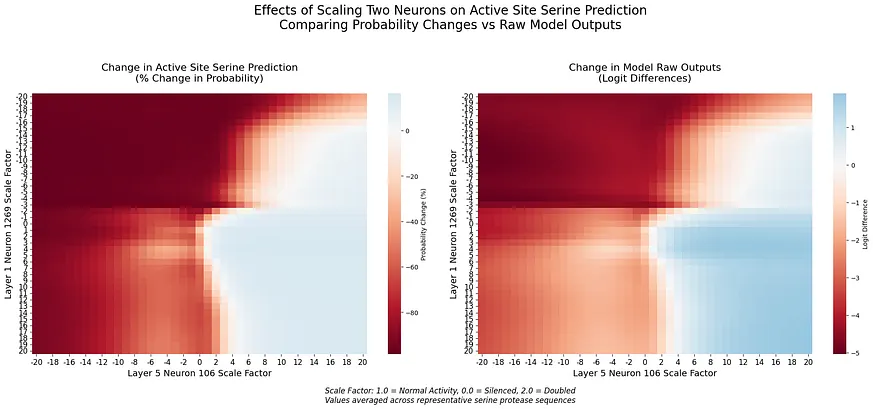
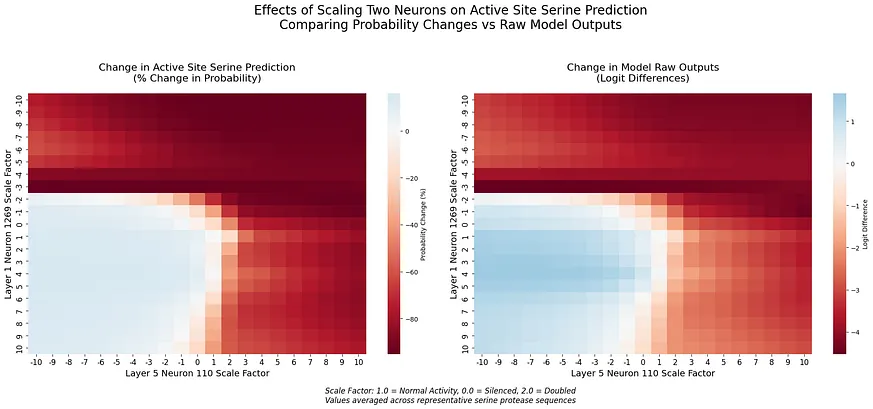
幾個關鍵模式浮現(詳情見[5]):不對稱影響:早期層神經元1269對第5層效果的調節強度明顯高於反之亦然。過渡邊界:當兩個神經元從正向縮放轉變為負向縮放時,預測會出現劇烈變化。這些發現表明,ESM-2在不同處理深度上採用了不同的編碼策略。雖然神經元1269作為一個早期且精確的特徵檢測器,第5層電路似乎則在更廣泛的序列上下文中整合了這些資訊。
蛋白質語言模型如何捕捉催化活性:一個令人著迷的發現
我們的主要發現——催化活性是透過一個在早期層次運作的精確檢測器,結合後期電路來編碼——暗示了蛋白質語言模型運作的一些迷人之處。儘管這些模型從大量序列資料中學習,並沒有明確的生物化學知識,但它們似乎能夠在不同處理深度下組織和編碼有意義的生物特徵,其表現與基於文字的語言模型驚人地相似 [6]。協同跨層計算來檢測催化位點的發現為我們的使命提供了強有力的證據。在 Reticular,我們正在努力使蛋白質設計變得更具可控性和可解釋性。尋找如此明確而簡潔的重要生物特徵表示,是一個令人鼓舞的第一步。仍然有許多工作要做。蛋白質語言模型:從催化性絲氨酸到更複雜的生物特性
我們對催化性絲氨酸的研究讓我們在理解蛋白質語言模型方面取得了一定的基礎,但我們也意識到存在的限制和未解決的問題。我們的分析集中於 ESM-2 的最小變體(8M 引數),並在一個簡單的二元性質中識別出層特定機制。驗證過程依賴於成熟的生化真實資料,而我們的探測方法可能會忽略更為分散的表徵。大多數生物特性並不像催化性絲氨酸那樣整潔地組織在各層之間。那麼,模型如何編碼更為混亂的特徵,例如:結合親和力(連續光譜)、熱穩定性(源自全域性結構)、構象變化(動態特性)呢?我們對模型規模如何影響這些表徵,以及不同架構之間如何比較,幾乎才開始探討皮毛。機制可解釋性:解鎖蛋白質工程的未來
我們正在積極尋求合作,以擴充套件這項工作:與您合作或設計蛋白質?我們希望能部署我們的技術,助您更快達成里程碑。請在 nithin [at] reticular.ai 預約聊天。如果您是機制可解釋性(Mechanistic Interpretability)研究者,對生物應用感興趣,我們擁有計算資源及有趣的問題可供探討。隨時聯絡 nithin [at] reticular.ai — 我們期待探索如何透過機制可解釋性,使生物模型更可靠且可控。這一特性接近於稀疏探測案例研究中的語義範例:像是複合詞,其意義依賴於區域性上下文;像是程式語言檢測,它需要更廣泛的結構理解;還有如語法特徵,則提供了明確無誤的事實基礎。
在當前蛋白質工程領域中,「機制可解釋性」已然成為熱門話題。傳統的機器學習模型雖能預測蛋白質功能,但常缺乏足夠的可解釋性,導致這些模型難以被理解和信任。而機制可解釋性專注於揭示模型做出預測的內部運作方式,使研究人員能夠深入了解蛋白質之間的結構、功能及演化關係。
關注此議題的不僅有蛋白質工程研究人員,也包括藥物開發者以及對蛋白質結構和功能感興趣的人士。他們希望透過理解模型預測背後的機制來提升其可靠性和控制能力,加速藥物研發及蛋白質設計流程。
最新趨勢顯示,圖神經網路(GNN)因具備處理圖狀資料之優勢,正逐漸成為該領域的重要工具。將其與機制可解釋性結合,可以加深對蛋白質結構與功能之間關係的分析,推動藥物發現及設計進展。多尺度模型(Multi-scale Models)也日益受到重視,此類模型考量了從原子細節到整體結構等多個層面,為機制可解釋性提供了一個全面而扎實的資料基礎。同時,視覺化工具亦在不斷演進,以便讓研究者直觀地觀察到模型如何做出預測並進一步分析其運作原理。
Reticular.ai 的技術獨具特色,不僅在於其強大的計算能力,更在於它所提供精準且深入解析生物系統的方法論。我們期待攜手共創未來,把握新技術帶來的新契機。
神經元1269在生物資料分析中的關鍵角色
這一點在生物資料中尤其明顯,因為訓練集和測試集之間的序列相似性可能導致效能指標虛高。我們特別注意確保我們的測試集包含真正與訓練資料低相似度的序列。為了檢查神經元識別的一致性,我們對四種方法進行了重疊分析:邏輯回歸、互資訊、均值差異和支援向量機 (SVM)。神經元1269在所有方法和k值下始終被100%識別,這表明其具有基本作用。相比之下,其他神經元(例如207和818)則顯示出不一致的識別,其中沒有其他神經元在各方法中的一致性超過50%。統計驗證突顯了神經元1269的重要性。在催化位點方面,其效應大小強勁(Cohen′s d = 8.523,針對負縮放),且分佈發生顯著變化(KS統計量 = 1.000,p = 0.029)。而對於非催化位點,其效應微弱(Cohen′s d = 0.099),且未觀察到顯著變化(KS統計量 = 0.062,p > 0.99)。分析跨層互動揭示了當神經元 1269 與神經元 110 和 106 互動時的不同模式。與神經元 110 的互動中,當兩個神經元的效應均為負向縮放時,觀察到強烈的抑制效應,這創造了一個明確的決策邊界於中性啟用周圍,並突顯出非線性效應,特別是在機率空間中。相比之下,與神經元 106 的互動則顯示出更為漸進的過渡,其補充性的啟用模式暗示了合作計算。在每種互動中,logit 空間和機率空間之間出現了不同的模式。
這些互動符合第五層神經元的原始特徵,其中神經元 106 和 110 對絲氨酸預測具有相反的影響:神經元 106 與絲氨酸預測呈正相關,而神經元 110 則呈負相關。神經元 1269 的互動反映了這種互補性,與神經元 110(對絲氨酸預測產生抑制作用)之間創造出明顯的轉變,以及與神經元 106(增強絲氨酸預測)之間平滑的調節。這些在跨層次互動中保留相對角色的情況,暗示著該互補性對模型處理催化位點而言是根本性的。
蛋白質語言模型的層級組織:突破傳統語言模型的框架
在對於架構比較的一則說明中,Gurnee 等人發現在 Pythia 模型的中間層中主要存在可解釋的特徵(該模型擁有 6 至 32 層),而我們在 ESM-2 的研究結果則顯示出不同的模式:第 1 層有一個精確的特徵檢測器,而在其 6 層架構中的第 5 層接近輸出的地方有一個整合電路。這種差異相當顯著——儘管我們使用了類似的探測方法,但其層級組織與傳統語言模型看起來卻截然不同。可解釋特徵同時存在於網路的兩端,而非集中於中間層,這可能反映了蛋白質語言模型在資訊組織上的根本性差異,或者與 ESM-2 的小規模及不同架構有所關聯。觀察到跨層互動作用進一步暗示蛋白質模型可能採用了獨特的計算策略,相較於文字模型而言更為複雜。**專案1:蛋白質語言模型的層級組織新發現**
傳統語言模型的特徵通常集中在中間層,但研究表明,ESM-2 模型中,第 1 層的精準特徵檢測器與第 5 層的整合電路協同工作,形成了獨特的層級組織方式。這反映了蛋白質語言模型可能採用與文字模型截然不同的資訊組織方式,為理解蛋白質序列和結構提供了全新的視角。
**專案2:跨層互動作用的深層意義**
ESM-2 模型中觀察到的跨層互動作用,表明蛋白質語言模型可能採用了比文字模型更復雜的計算策略。這也為探究不同層級之間的互動作用提供了新的方向,例如,可以研究不同層級的特徵如何在相互作用中共同塑造最終預測結果,以及這種跨層互動作用如何影響模型的泛化能力和魯棒性。
以下是針對不同稀疏度水平 (k) 在保留的測試集上,各方法的外部樣本 F1 分數,包括第 5 層的結果。
神經元分析:揭示催化性絲氨酸功能的關鍵神經元組合
幾個引人注目的發現浮現出來:大多數方法在僅使用 k=2 個神經元的情況下即可達到完美的 F1 分數 (1.000)。隨機選擇在所有 k 值中表現不佳,這驗證了我們的方法論。支援向量機 (SVM) 在單一神經元效能中顯示出最強的表現(0.667)。而在 k=2 之後,效能完全趨於平穩,這表明我們已找到最小的表示。我們的分析集中於第五層中的兩個關鍵神經元,這些神經元似乎編碼催化性絲氨酸功能。值得注意的是,一些方法識別到了替代神經元(例如,在邏輯回歸中,第287號神經元顯示出相反關係),但106–110這對組合在各種方法中顯示為最可靠。以下是它們的詳細統計資料:
第106號神經元作為主要特徵檢測器浮現出來:
- 在所有方法中一致被識別
- 大幅度平均啟用差異 (1.560)
- 強效應大小 (2.462)
- 正案例(8.153)與負案例(6.593)之間清晰分隔
第110號神經元似乎扮演支援角色:
經由多種方法(MI、平均差異、SVM)識別出來,效果大小為中等但一致(1.158)。不同類別之間的啟用差異較小但顯著。透過將神經元的啟用值從基線值的-10倍逐漸提升至+10倍,我們觀察到對模型的絲氨酸預測產生高度可控的影響。神經元106
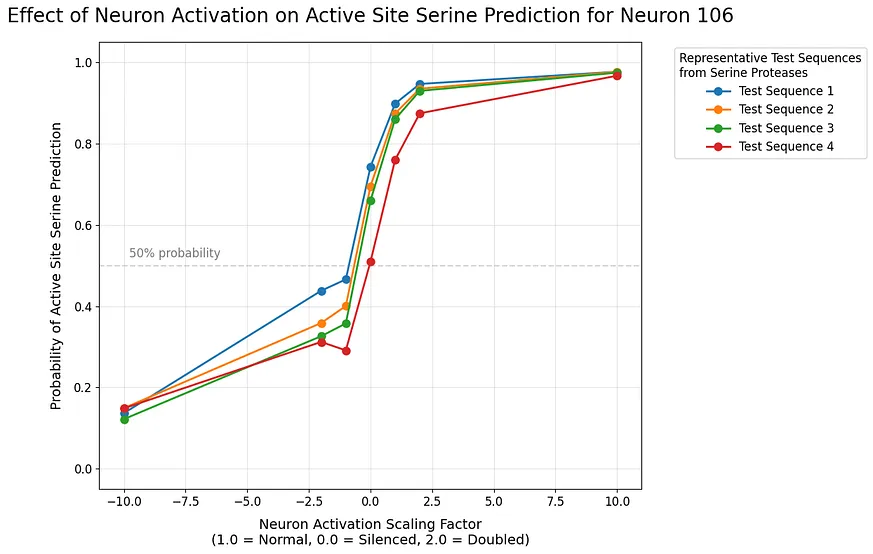
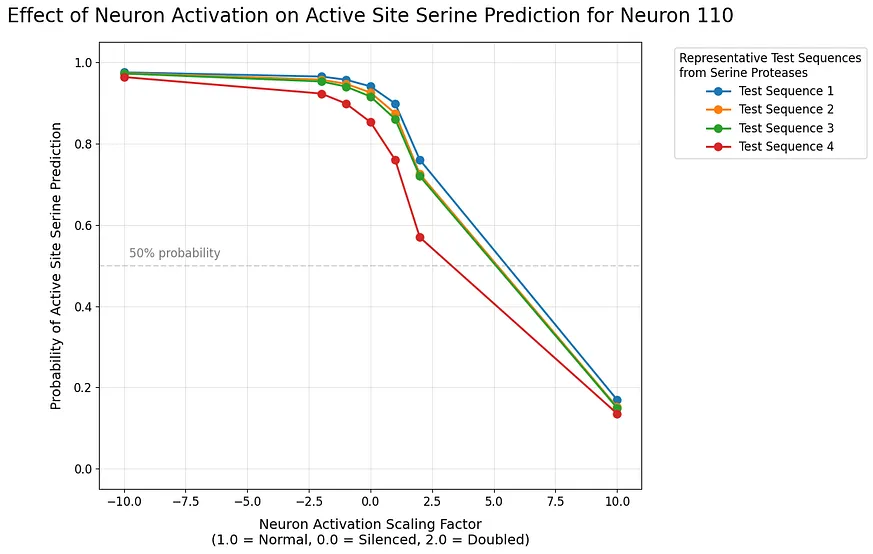
深度學習揭示神經元功能:催化絲氨酸與神經元 106、110 的互動關係
神經元 106 顯示出強烈的正相關性——不斷增加其活化能夠持續提升模型對催化絲氨酸的預測。這種關係在邏輯空間中表現得相當線性,暗示此神經元直接編碼了催化絲氨酸的功能。相較之下,神經元 110 的行為則更為複雜。儘管它通常與神經元 106 的效果相對立,但當其被下調時,其影響力比上調時更為明顯,這表明它可能扮演著調節角色。當兩者一起進行縮放時,我們觀察到既有協同增強又有相互抵消的現象:
協同增強:當兩個神經元以相容的方向進行縮放(即 106 上升、110 下降)時,我們看到對於絲氨酸預測信心的超加效應。
抵消:當兩者朝著相反方向縮放時,它們能有效地中和彼此的影響。
這段文字揭示了神經網路中特定神經元與特定功能之間的直接關聯性,並進一步描述了不同神經元之間的互動作用。這與當前神經科學領域的重要趨勢——「神經元功能解碼」密切相關。近年來,研究人員積極探索如何利用深度學習技術反向推導出神經元活動與特定認知功能之間的對應關係。本研究中所觀察到的線性關係,以及神經元 106 和 110 之間複雜且富有啟發性的互動作用,為該領域提供了新的證據和研究方向。因此,我們不禁思考:如何利用深度學習模型解碼腦內各類特殊功能?以及如何透過理解這些互動作用來掌握複雜認知功能背後運作原理?
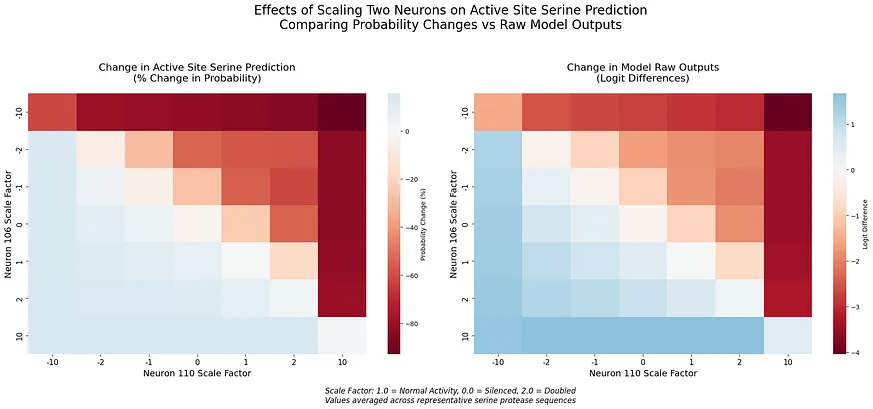
熱圖視覺化揭示了這些神經元之間如何相互調節的明確模式。為了驗證這些神經元是否特別編碼催化性絲氨酸,而非一般的絲氨酸,我們檢查了它們在不同絲氨酸族群中的影響。
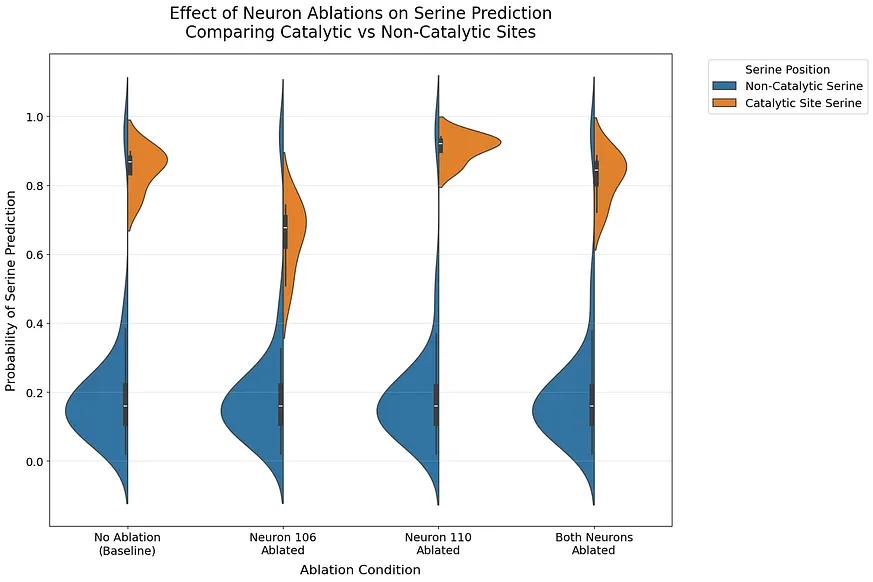
特定神經元調控催化性絲氨酸預測,揭示神經網路的精細結構
小提琴圖顯示出驚人的特異性:消融這些神經元對催化性絲氨酸的預測影響極大,而對非催化性絲氨酸的預測則幾乎沒有改變。當同時消融這兩種神經元時,影響最為明顯。我們進行了 Kolmogorov-Smirnov 測試以量化這些差異。消融神經元 106 與未消融相比顯示出顯著差異(KS = 1.000, p = 0.029),而消融神經元 110(KS = 0.750, p = 0.229)和同時消融兩個神經元(KS = 0.500, p = 0.771)則未顯示出顯著差異。針對非催化性絲氨酸,所有比較均表現出微小的效果(KS 統計量 < 0.031, p > 0.99),確認這些神經元專門調整於催化性絲氨酸。**專案1具體說明:特定神經元與催化性絲氨酸的精細調控關係:**這段研究結果揭示了特定神經元對於催化性絲氨酸預測的顯著影響,而對於非催化性絲氨酸則幾乎沒有影響。這說明瞭神經網路可能存在精細的結構,讓不同的神經元專注於處理不同型別的資訊,為我們理解神經網路如何處理複雜的生化資訊提供了新的視角。結合當前神經科學的發展趨勢,這項發現也為探討「神經元特異性」與「特定生化功能」之間的聯絡提供了新的研究方向。
**專案2具體說明:雙重神經元消融的協同效應:**研究顯示,單獨消融神經元106或110對催化性絲氨酸預測的影響有限,但當同時消融這兩種神經元時,影響效果顯著增強。這暗示了神經元之間存在複雜的互動作用,而單純分析單個神経員可能無法完整揭示其運作機制。此發現與目前「神経網路聯絡」研究方向相呼應,越來越多證據表明,不同 神經 元之間 的聯絡及其協同作用是實現複雜功能的重要基礎。因此,此項研究透過消融實驗提供的新證據,有助於未來深入探討 神經 網路 的運作機制。
參考來源
Nature 2024年重要的七項科技之一:蛋白質序列的深度學習模型
本文章介紹了Nature期刊中關於蛋白質序列的深度學習模型以及未來應用的重要性。蛋白質設計的應用從生物醫學到環境科學等各個領域解決問題方面具有巨大 ...
來源: Vocus深度學習為蛋白質科學開拓了下一個境地- Physics Today - 物理雙月刊
任何成功的蛋白質結構預測方法,包含新的深度學習模型,都需要仰賴生物學的觀點,而不是只有化學和物理學。 「蛋白質的序列不是隨機的。 它們是透過天擇提煉出來的。」 來 ...
來源: 物理雙月刊基于深度学习的蛋白质建模与设计
深度学习作为一种高效的数据特征提取方法,可以通过它对蛋白质数据进行建模,进而加入先验信息对蛋白质进行设计。故此基于深度学习的蛋白质设计就成为一个具有广阔前景的研究 ...
來源: 生物工程学报生物界的ChatGPT来了! 蛋白质语言模型ProGen助力特定功能 ...
受到基于深度学习的自然语言模型的成功启发,该研究团队开发了ProGen,这是一种蛋白质语言模型,在数百万个原始蛋白质序列上训练,可生成跨多个家族和功能的人造蛋白质。 ...
來源: 南京肽业生物科技有限公司AlphaFold3:當前最重要的突破與其對生命科學的影響
它們利用深度學習和演演算法最佳化,大幅提高了預測的準確性和速度,這些模型已解開無數蛋白質的結構,包括以往難以捉摸的膜蛋白,有效推動生物醫學研究 ...
來源: Imaging CoE深度学习模型与物理知识结合的蛋白质设计 - 酶有科技
蛋白质语言模型(pLM)通过学习氨基酸序列的共进化信息,可以预测序列的下一个氨基酸。我们使用核酸酶序列的blast结果对蛋白质语言模型使用LORA微调得到可以生成多样核酸 ...
來源: mayootech.com科學家開發新的深度學習模型,在10分鐘內就可完成蛋白質結構預測
研究團隊使用不連續的蛋白質片段來訓練模型,這些片段具有260個獨特的氨基酸元素,藉由利用PyTorch深度學習框架,來推斷蛋白質的化學組成以及摺疊結構。
來源: iThome【生成式AI醫級戰場1】全球10獨角獸創新局快速生成蛋白質新藥
其中,最強大的AI模型RFdiffusion是一款結合結構預測和生成式擴散模型的創新AI系統,它可以透過深度學習生成與自然界中任何已知蛋白質不同的新蛋白。利用 ...
來源: 國家新創獎
 全部
全部 康健
康健
相關討論